Classificação Molecular do Carcinoma da Mama
Guilherme Portela Coelho (CRM 23795)
Biologia Molecular
Biologia molecular é o segmento da genética que estuda os ácidos nucleicos (DNA e RNA), bem como outras moléculas ou alterações que influenciem nas suas funções.
Este estudo corresponde a "área laboratorial" da genética. Podendo avaliar tanto as sequências das bases nitrogenadas (adenina, timina, guanina, citosina e uracila) que compõem o material genético, bem como, a sua expressão.
Entende-se por expressão gênica a tradução da sequência de nucleotídeos de um segmento da molécula de DNA para RNA, este último com capacidade de carregar a informação para fora do núcleo celular e influenciar na síntese de proteínas.
Em última análise, quem executa o trabalho derivado das instruções (informações) genéticas são as proteínas, que são codificadas pelos genes.
De uma forma sintética e simplificada, para um funcionamento saudável das células e de um organismo, é importante a integridade dos mecanismos de armazenamento (DNA) de informação genética, de transcrição e transporte desta informação (RNA) e de tradução da mesma para proteínas (síntese proteica).
Alterações ou disfunções que podem participar de processos patológicos são diversas quando se fala em biologia molecular. Estas alterações podem ocorrer na sequência de bases do DNA, na expressão (transcrição para RNA) ou na tradução do código (síntese proteica).
Na sequência do DNA podemos ter ganhos e perdas de informação (bases nitrogenadas), trocas de bases, mudança de local de determinada sequência (translocação) e aumento do número de cópias de um determinado gene sem necessária alteração na sequência de bases. Todas têm potencial de produzir proteínas disfuncionais (mais ou menos ativas) ou alteração na expressão de outros genes.
Como se as alterações de sequência e quantidade de DNA não fossem suficientes para explicar uma grande gama de situações patológicas, a função dos genes ainda sofre influência de mecanismos epigenéticos.
Epigenética é a área do conhecimento que trata de alterações nas funções dos genes, mas por influência de interações que não tem diretamente relação com as sequências (de bases nitrogenadas) destes.
Interação do DNA com histonas, metilação de bases nitrogenadas e os microRNAs são exemplos de situações/fatores que influenciam em como, quando e porque os genes se expressam e se traduzem em proteínas.
Classificação Molecular
Por classificação molecular entende-se o agrupamento de casos de neoplasias de acordo com características comuns de:(1)
- a) mutações no DNA celular (alterações na(s) sequência(s), incluindo aumento de número de cópias de genes);
E/OU
- b) padrões de expressão genica, ou seja, quais genes estão sendo mais transcritos ou menos transcritos para RNA, independentemente de serem mutados ou não;
E/OU
- c) presença de eventos epigenéticos patológicos (área mais incipiente em comparação às anteriores);
Métodos (mais comuns) de estudo molecular.
Para pesquisar alterações na sequência de genes, podem-se utilizar diversos métodos diretos ou indiretos. Um exemplo indireto é assumir alteração genética a partir de defeito na proteína correspondente, contudo, este tipo de avaliação foge ao escopo deste capítulo.
As técnicas mais comuns para detecção de alterações genéticas, consequentemente, para classificação dos tumores por esta perspectiva são a reação da polimerase em cadeia (PCR) e o sequenciamento gênico.
Bastante resumidamente, pode-se dizer que a técnica de PCR pode identificar variações genéticas específicas, previamente conhecidas, e é custo-efetiva quando não necessitamos examinar muitos locais diferentes do mesmo gene. Assim, serve majoritariamente para estudo de genes que apresentam um padrão mais "monótono" de alterações, ou seja, costumam apresentar mutações quase sempre nos mesmos locais da sequência e muitas vezes trocando de forma também monótona uma base por outra. Um exemplo é a mutação V600E do gene BRAF, onde há troca de uma mesma base por outra no mesmo ponto da molécula de DNA.
Também por PCR, sobretudo com o advento da reação / mensuração em tempo real (RT-PCR ou PCR quantitativo), pode-se avaliar a expressão genica. Nesta modalidade, descobre-se o que a célula estaria "fazendo" extraindo dela seu RNA, ou seja, descobre-se apenas quais genes estão sendo expressos (transcritos de DNA para RNA).
Para tal estudo, extrai-se o RNA e, por conta da sua labilidade, gera-se uma molécula complementar de DNA em laboratório (cDNA, que é mais estável) que carrega consigo a informação análoga ao RNA extraído, para então realizar-se a reação de PCR e sua “leitura".
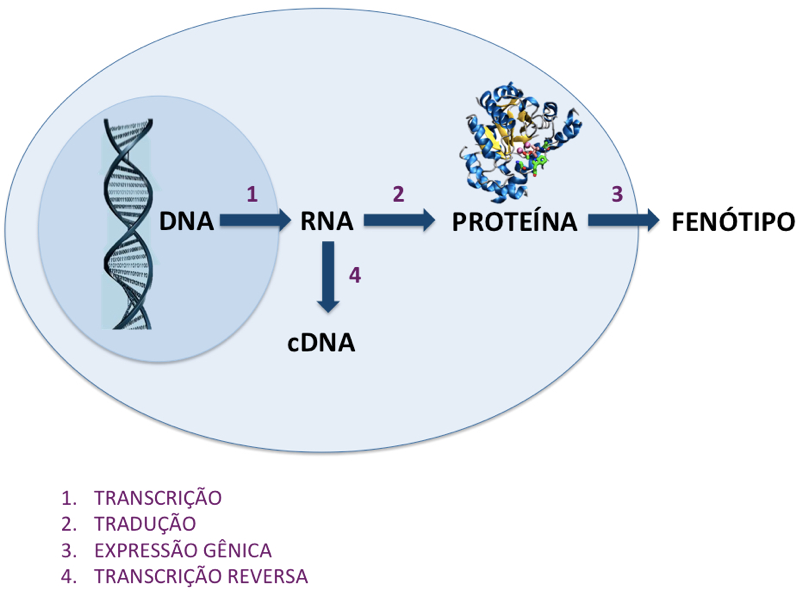
Figura 1:Modelo demonstrativo da codificação proteica presente no nosso genoma (horizontal). cDNA (c=complementar) é a forma de estabilizar a informação extraída do RNA presente (extraído) de um determinado tecido nos estudos de expressão genica.
Os ensaios que geraram a absoluta maioria dos dados de biologia molecular referente ao diferentes tipos de carcinomas de mama são provenientes de estudos de RNA (expressão genica), comparando tumores em relação a quais genes estão sendo transcritos (ou ativos).
Quando o padrão de mutação genética não é "monótono", ou inclui muitas regiões diferentes de um gene, utiliza-se a reação de sequenciamento. Esta consiste em detectar a sequência de bases e determinado gene independentemente dela ser conhecida ou previsível.
Um exemplo são os genes BRCA 1/2 (em seu estado normal, protegem a célula epitelial mamária da transformação em câncer). No caso destes genes, alterações na sequência que podem levar à disfunção já foram detectadas em diversos pontos do mesmo, inviabilizando na prática a pesquisa a partir de sondas (para reação de PCR) contra todas as possibilidades conhecidas como patológicas.
O sequenciamento clássico é chamado de método de Sanger e, recentemente, tem sido substituído por sequenciamento de nova geração (NGS) que gera maior volume de dados em menor tempo e a um custo menor.
Esta última técnica é a recomendada, salvo em situações onde se queira pesquisar deleções ou adições de grande volume de DNA pois a fragmentação do DNA inerente a este método não parece ser confiável (até hoje) para este tipo de situação. Por exemplo: após resultado negativo para pesquisa de mutação de BRCA 1/2 por NGS, deve-se confirmar a negatividade por MLPA (tecnologia de Sanger) pois o NGS não detecta grandes ganhos ou perdas de material genético.
Por que classificar? Um breve histórico.
A noção de que o câncer é uma doença única há muito tempo já foi abandonada. Hoje sabemos que, epidemiologicamente e evolutivamente, este grupo de doenças é extremamenteheterogêneo.
Não é aceitável abordar um carcinoma de pulmão da mesma forma que um carcinoma da próstata, mesmo que ambos possam ser chamados de adenocarcinoma.
Ainda assim, saber apenas o órgão de origem e a histogênese é informação demasiadamente superficial para estabelecerem-se os cuidados necessários para um paciente com câncer.
Para escolher o melhor tratamento dispomos de uma série de protocolos específicos de manejo da doença, baseados em testes científicos e que levam em consideração o potencial de agressividade de cada câncer, bem como o estágio de evolução do mesmo (derivado do tempo do seu aparecimento até o momento do diagnóstico).
Tudo isso evoluiu através do estudo dos tumores, submetidos a diferentes métodos de observação, desde a visualização direta, passando pela microscopia, reações imunológicas e alcançando a biologia molecular.
Isoladamente, o carcinoma de mama é uma doença tão heterogênea quanto acreditávamos serem todos os tipos de câncer séculos atrás.
Passando pela forma e arranjos das células (tipos histológicos e graus histológicos), detecção de expressão de proteínas (imuno-histoquímica para receptores hormonais e HER2/neu e culminando na pesquisa de sequências genéticas alteradas e expressão gênica destes tumores, hoje é possível classificá-los mais acuradamente do que nunca em relação ao seu comportamento e saber, ainda, se respondem a tratamentos específicos, bem como municiar os cientistas com ferramentas poderosas para seguir avançando no entendimento da doença.
Histórico: origens e marcadores clássicos
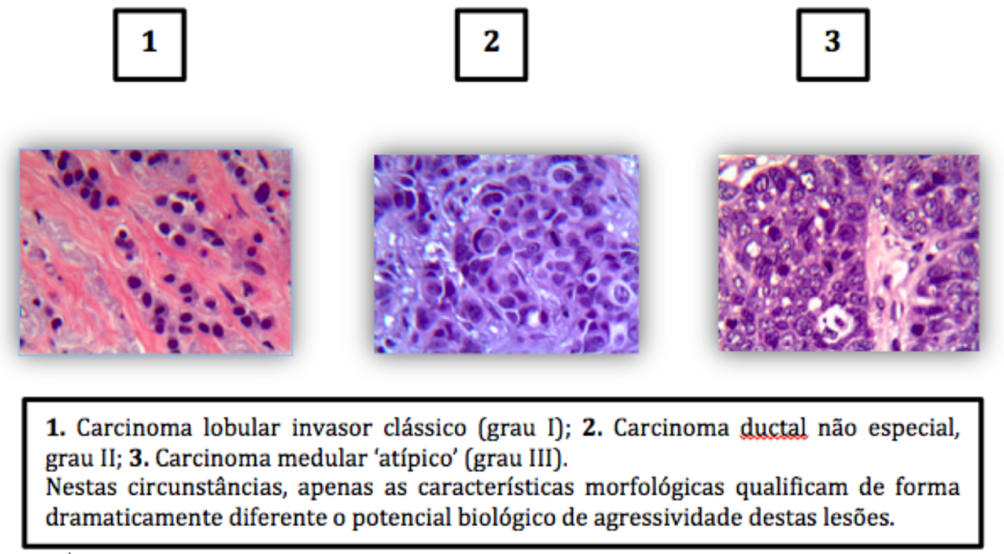
O que chamamos de morfologia é o conjunto de achados aferidos e estudados por observação da forma das células e dos tecidos. Repare nestas diferenças tomando como modelo as três imagens abaixo, todas de carcinomas da mama, mas com diferentes tipos histológicos.
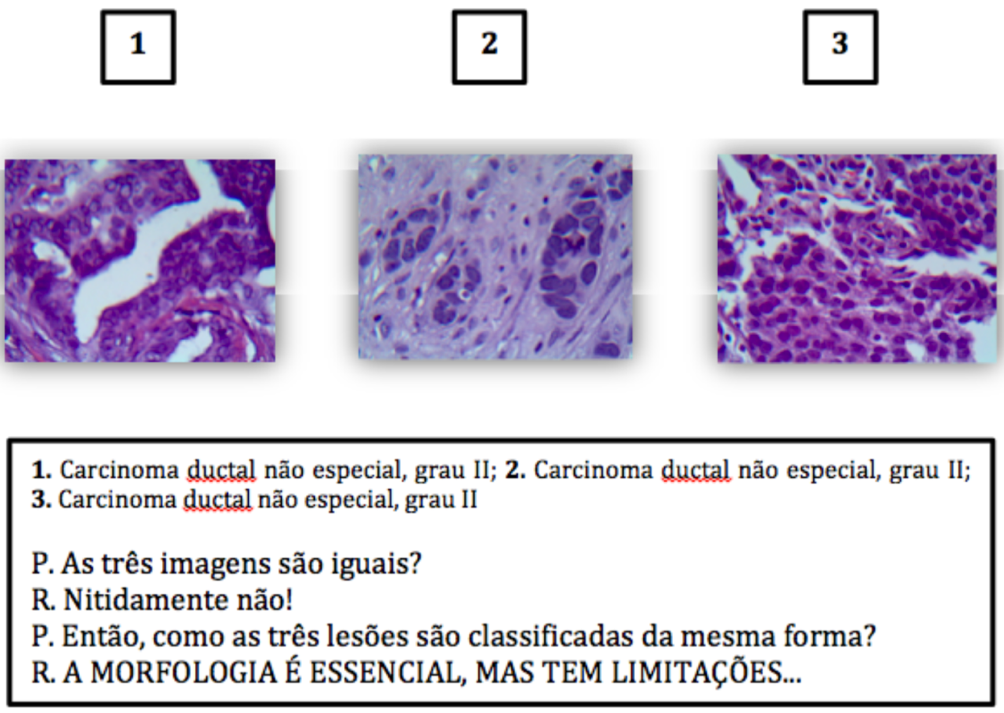
Entretanto, infelizmente, tumores de mesmo tipo histológico, mesma dimensão, mesmo grau histológico e mesmo estadiamento podem apresentar evoluções diferentes:
Além da morfologia, já utilizados há bastante tempo e ainda muito úteis, são o perfil de expressão de receptores hormonais (estrogênio e progesterona) e a proteína her2/neu. Atualmente dispomos de métodos simples de detecção, que nos permitem avaliar se um determinado carcinoma mamário expressa estas características, que podem ser utilizadas como:
- a) MARCADORES PROGNÓSTICOS INDEPENDENTES DOS FATORES MORFOLÓGICOS E CLÍNICOS CLÁSSICOS: o ‘status’ positivo para receptores hormonais significa que a neoplasia se assemelha ao tecido normal neste aspecto, sendo “melhor diferenciada funcionalmente”.
Cerca de 80% dos carcinomas invasores da mama expressam receptores para estrógeno. Já os receptores de progesterona estão expressos em cerca de 60-70% dos casos e indicam que a via para o receptor de estrógeno está intacta e funcionando.(13)
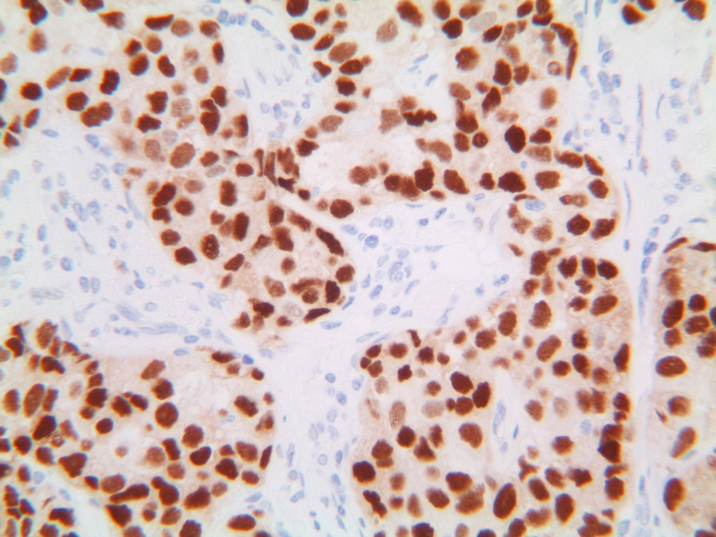
Figura 4:A positividade para receptores estrogênicos está demonstrada pela coloração marrom no núcleo das células deste carcinoma - imuno-histoquiímica (IHQ) com anticorpo anti-RE, 400x
- b) MARCADORES PREDITIVOS PARA UMA RESPOSTA TERAPÊUTICA MAIS ESPECÍFICA DO QUE OS FÁRMACOS TRADICIONAIS: a proteína Her2 localiza-se na membrana celular e, apesar de não ter um ‘ligante’ específico conhecido, é capaz de ativar uma cascata de sinalização que encaminha a célula para multiplicar-se; há drogas disponíveis que ligam-se a esta proteína (anticorpo monoclonal), bloqueando a sua ativação e promovendo a destruição celular, através da ativação imunológica do organismo.
Da mesma forma, o receptor de estrogênio desencadeia proliferação celular quando ligado ao hormônio, havendo terapia que bloqueia esta ligação. Estes casos tem valor preditivo positivo para resposta ao tratamento de bloqueio hormonal ou Inibidores da Aromatase (enzima responsável pela conversão do precursor do estrogênio). A proteína Her2 está super-expressa em cerca de 15% dos carcinoma primários da mama. Em 2007 foi considerado parâmetro preditivo de resposta terapêutica pela Sociedade Americana de Oncologia Clínica.
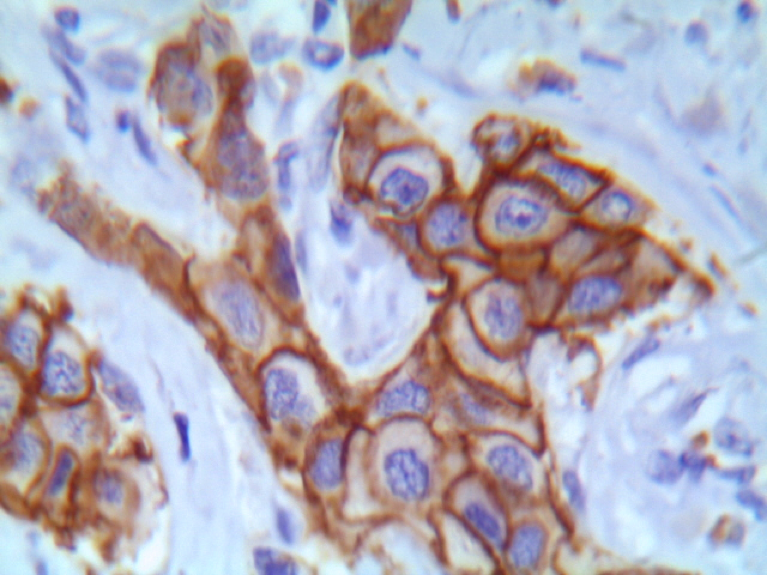
Figura 5:Escore 3+ na imunorreatividade para Her2/neu
Há um grupo de carcinomas que provavelmente não represente uma única e distinta “entidade” patológica, mas sim o conjunto de lesões não classificáveis pelos critérios que enquadram os tipos anteriormente citados. Igualmente, como grupo, tendem a apresentar mau prognóstico (considerando a história natural da doença). São chamados de “triplo-negativos”, ou seja, negativos para receptores hormonais (estrogênio e progesterona) e também para HER2/neu.
Por serem negativos para os marcadores preditos de terapia direcionada (biomarcadores hormonais e Her2), não costumam ser alvo de terapia específica, contudo por apresentarem altos índices de proliferação celular (ki67) em comparação com os outros tipos, apresentam boa resposta (eventualmente completa) à quimioterapia convencional.
Perfis de expressão genica - A Classificação Molecular
Com o surgimento e desenvolvimento das técnicas de estudo da expressão de genes a partir da detecção do RNA presente nas células, iniciou-se a classificação das doenças, bem como subclassificação dos carcinomas de mama, de acordo com a sua “assinatura molecular”.
Como a maneira de um determinado gene (DNA) tem de se expressar é através do seu respectivo RNA, pode-se deduzir quais genes são importantes para cada evento que caracterize o câncer (proliferação, invasão, metástase), comparando-se tecidos normais com carcinomas que apresentem os diferentes comportamentos biológicos. Também, desta forma, podem ser comparados diferentes carcinomas entre si.
Este campo (relativamente novo) de estudo, já encontra aplicação prática em contextos clínicos selecionados, por exemplo, em um determinado carcinoma que pode ser biologicamente agressivo e estar em estágio inicial pelos métodos de estadiamento utilizados. Nesta circunstância, mesmo com estadiamento apontando doença inicial e, eventualmente, com os fatores prognósticos mais antigos “limítrofes”, se justificaria uma terapêutica mais intensa para bloquear a progressão da doença.
Van’t Veer et al e van de Vijver et al (14, 15) foram os primeiros a aplicar a análise de expressão de genes para definir subgrupos de pacientes com carcinoma da mama com maior probabilidade de desenvolver metástase. Em 2003 uma análise preliminar de 70 genes (obtidos após uma triagem de milhares de genes expressos nos tecidos de tumores da mama!) mostrou ter valor prognóstico superior ao painel de critérios prognósticos clássicos utilizados pelo consenso de St. Gallen à época.
Com base na expressão destes genes, surgiram testes comercialmente disponíveis. Estes testes dividem as lesões de acordo com “índices de recorrência”, ou classificam o “tipo molecular propriamente dito” refletindo a probabilidade de um carcinoma com determinadas características de recidivar local ou sistemicamente.
Os resultados dos estudos baseiam-se nos achados que demonstram que os carcinoma da mama tendem a agrupar-se em padrões predominantes com relação ao RNA expresso nas suas células. Estes padrões definem grupos biologicamente distintos com ênfase em genes responsáveis pela proliferação celular, diferenciação celular (de célula imatura para madura), capacidade de infiltração / disseminação e sobrevivência.
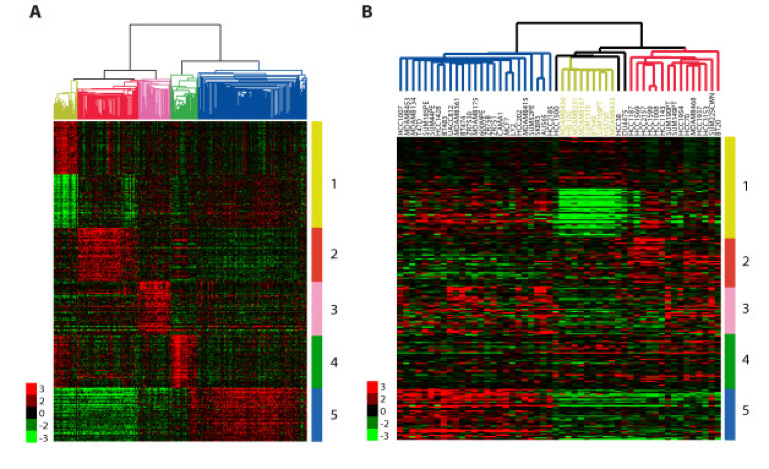
Figura 6:Os padrões demonstrados através de diagramas que apontam, através de cores, os genes mais expressos (mais transcritos para RNA), menos expressos e nulos, em relação a um referencial (célula não neoplásica). Imagens como esta são cada vez mais frequentas não apenas em oncologia mamária. Resulta de agrupamento hierárquico dos muitos (eventualmente milhares) de pacientes estudados em relação a expressão genica (RNA) nas células tumorais. Cada pequeno local colorido representa a expressão de um dentre centenas de genes diferentes (selecionado entre milhares após estudos
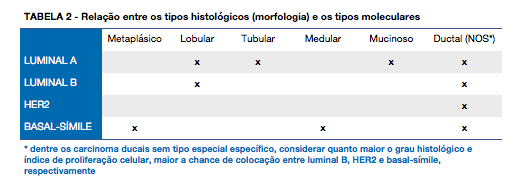
Resumidamente, e comparando-se com o conhecimento prévio de morfologia e imuno-histoquímica, os tipos moleculares de carcinomas de mama que são aceitos pela maioria dos especialistas são (1):
- a) LUMINAL ‘A’: carcinoma que expressa receptores hormonais, não tem amplificação do gene ERBB2 (código da proteína Her2-Neu) e apresenta baixo índice de proliferação celular aferido pelo marcador ‘ki67’; este tipo de lesão (luminal) se origina das células que revestem o lúmen dos ductos/lóbulos mamários e tem o melhor prognóstico dentro desta classificação ‘molecular’ dos carcinomas da mama.
- b) LUMINAL ‘B’: carcinoma igualmente originário do epitélio ductal, também expressa receptores hormonais, contudo o índice de proliferação celular (ki67) é maior ou igual a 15%. São negativos para Her2.
- c) HER2 amplificado (ou enriquecido): carcinoma mamário que se caracteriza por apresentar amplificação (aumento do número de cópias) do gene ERBB2. Costumeiramente, estas lesõessão negativas para receptores hormonais. Apresentam grande potencial de agressividade biológica, porém, têm resposta clínica quando tratados com anticorpo anti-Her2 ou outros fármacos que bloqueiam a cascata de eventos relacionadas a este receptor no interior das células. Supostamente, a origem também é nas células luminais dos ductos mamá
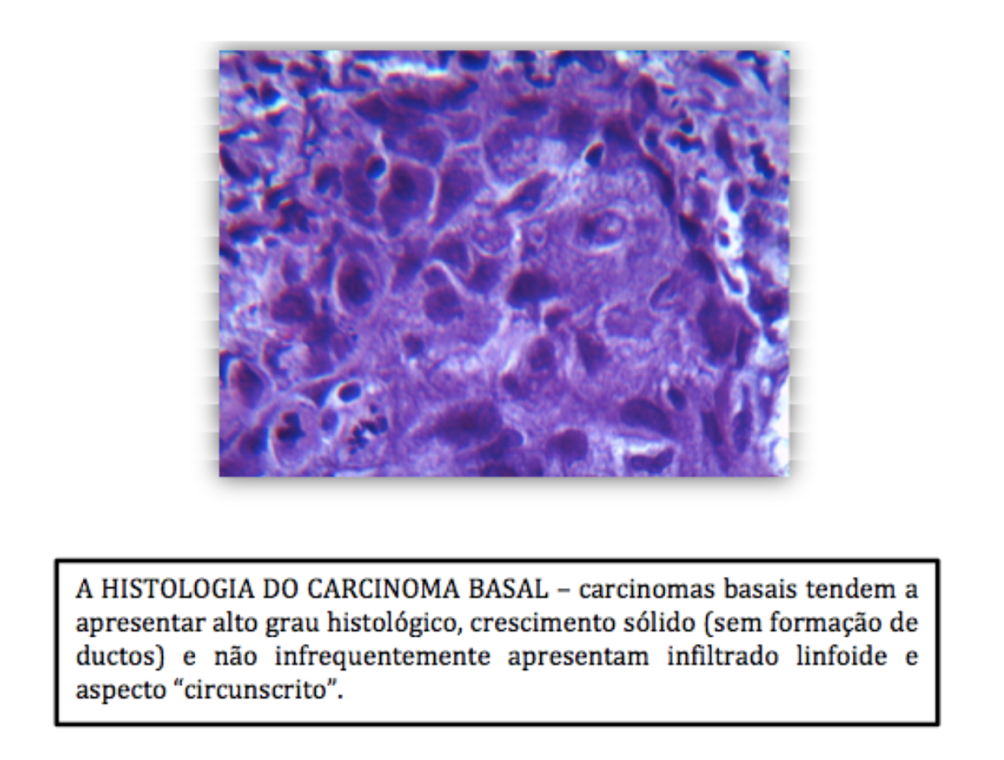
- d) BASAL-SÍMILE (“BASAL-LIKE”): este tipo de tumor supostamente se origina das células “basais” dos ductos mamários em virtude da expressão das citoqueratinas 5 e 6 (assim como as células não neoplásicas da localização basal dos ductos). São negativos para amplificação do ERBB2, não expressam receptores hormonais e são, também, positivos para expressão de EGFR (receptor do fator de crescimento do epitélio). São biologicamente agressivos, contudo, têm alta taxa de resposta a agentes quimioterápicos convencionais. O índice de proliferação celular, medido por “ki67" é comumente elevado, podendo ser superior a 90% das células.
O carcinoma de tipo molecular basal-símile é comumente confundido com os tumores classificados como “triplo negativo” pela imuno-histoquímica. Esta comparação não é injustificada, uma vez que praticamente todos os carcinoma basais-símile não expressam consideravelmente receptores hormonais nem hiper-expressam HER2. Contudo, nem todos os casos triplo-negativos apresentam perfil molecular semelhante às células basais da mama, tampouco expressão imuno-histoquímica de citoqueratinas 5/6, egfr ou outros marcadores proteicos de células basais.
A busca por uma categorização dos casos “triplo-negativos” é um tema de pesquisa instigante. Mais recentemente, foram propostas mais classificações: I) “claudin-low” ou carcinoma pobre em claudina, II) carcinoma tecido-normal-símile (“normal-like”), III) carcinoma triplo-negativo com expressão de receptores de androgênio (RA)(16), IV) carcinoma imunomodulatório (quiçá alguns sejam possível alvo para imunoterapia), e V) carcinoma com transição epitelial-mesenquimal. A lista é crescente.
As claudinas são moléculas de adesão intercelular cuja expressão pode estar perdida em alguns casos de carcinoma mamário, sobretudo do tipo “triplo-negativo”. Destas proteínas, as que são avaliadas nos carcinoma mamários com mais frequência são as de número “1”,”3”,”4” e “7”.
Para alguns casos remanescentes, ou seja, aqueles onde não se consegue classificar de acordo com os tipos moleculares conhecidos, propôs-se, de forma controversa, o rótulo de normal-símile, ou seja, semelhante ao tecido normal na expressão genica. Inicialmente parece paradoxal o fato de que estes não expressam receptores hormonais, contudo, o tecido mamário normal apresenta expressão menos intensa, em menor proporção de células e descontínua no tempo (ciclos hormonais) para RE e RP. Então, esta expressão costuma ser, em um grande número de vezes, menos intensa e frequente (em proporção de células) do que nos carcinoma “luminais”.
E quanto à epidemiologia? Neste campo, considerando-se faixas etárias de incidência e inferindo-se que os agentes genéticos hereditários e estímulos ambientais possam identificar tipos distintos de câncer da mama, podemos observar que os picos de diagnóstico de câncer da mama demonstram que por um lado o carcinoma ‘basal-símile’ e por outro o conjunto dos demais ‘tipos moleculares’, se comportam como ‘tipos etiológicos’ distintos.(17)
Nesta perspectiva, os carcinomas ‘basais-símile’ e muito provavelmente os triplos-negativos representariam um grupo ligado mais fortemente à herança genética, aparecendo mais cedo no ciclo de vida, enquanto que os demais tenderiam a se manifestar mais tardiamente e possivelmente estariam mais ligados aos efeitos de estimulação crônica do tecido por hormônios ou exposição a carcinógenos ambientais.(17)
Apesar das evidências epidemiológicas, há muitas arestas remanescentes, sobretudo neste grupo dos ‘não basais’, por exemplo: qual a real acurácia da morfologia e da imuno-histoquímica juntas em diferenciar os carcinomas entre bom prognóstico e mau prognóstico quando tratamos dos tipos ‘luminais’ (A e B)? Esta é uma questão crucial em determinados contextos clínicos onde se deve decidir pela utilização ou não de tratamento sistêmico (quimioterapia), principalmente em pacientes pós-menopausa ou com tumores em estágios iniciais.(18).
Correlação: painéis prognósticos versus morfologia
Os tipos histológicos que geralmente recebem baixo grau histológico, usualmente se classificam como de tipo luminal A, já as variantes menos diferenciadas como “basal-like”.
O maior problema, no que se refere a heterogeneidade, é o que chamamos de ductal não especial (“not otherwise specified”ou NOS), que é uma categoria de exclusão, representando mais de 90% das lesões diagnosticadas. As classificações moleculares/genéticas devem suprir esta deficiência das classificações morfológicas.

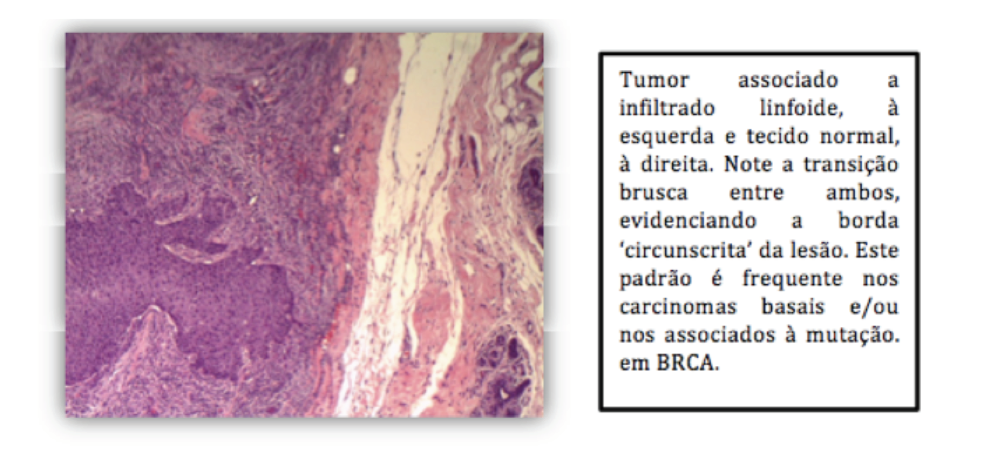
A presença de padrão expansivo em bordas tumorais, infiltrado linfoide, alto grau histológico e necrose são características morfológicas associadas aos casos de carcinoma familiar, curiosamente, boa parte destes tumores também são associados ao fenótipo “basal-like” e também a existência de mutação dos genes BRCA 1/2 associa-se com mais força a esta morfologia.(xx) OS BRCA’s (‘BR’east ‘CA’ncer) são genes responsáveis pelos chamados casos ‘familiares’ de carcinoma mamário (herança autossômica dominante). Estão associados ao aparecimento de câncer em idade mais precoce. Não são utilizados com frequência para classificar os tumores em virtude do alto custo do exame (não se trata de apenas um tipo de mutação, mas uma variedade grande em um único gene que precisa ser completamente sequenciado para avaliar se há ou não alteração).
Referências Bibliográficas
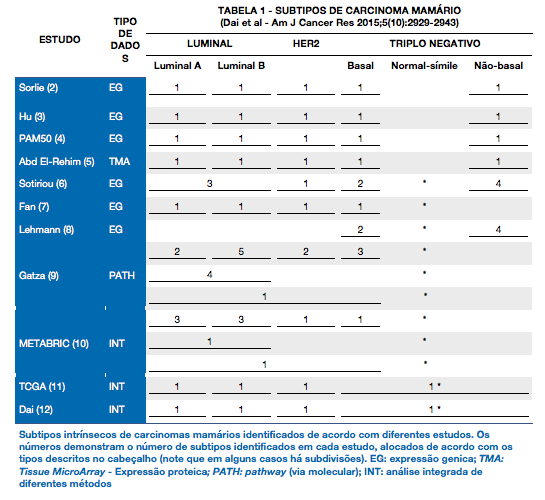
- Xiaofeng Dai, Ting Li, Zhonghu Bai, Yankun Yang, Xiuxia Liu, Jinling Zhan, Bozhi Shi Breast cancer intrinsic subtype classi cation, clinical use and future trends. Am J Cancer Res 2015;5(10):2929-2943
- Sørlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lønning PE, Brown PO, Børresen-Dale A and Botstein D. Re- peated observation of breast tumor subtypes in independent gene expression data sets. PNAS 2003; 100: 8418-8423.
- Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, Nobel A, Parker J, Ewend MG, Sawyer LR, Wu J, Liu Y, Nanda R, Tretiakova M, Ruiz Orrico A, Dreher D, Palazzo JP, Perreard L, Nelson E, Mone M, Hansen H, Mullins M, Quackenbush JF, Ellis MJ, Olopade OI, Bernard PS and Perou CM. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics 2006; 7: 96.
- Dowsett M, Sestak I, Lopez-Knowles E, Sidhu K, Dunbier AK, Cowens JW, Ferree S, Storhoff J, Schaper C and Cuzick J. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant re- currence after endocrine therapy. J Clin Oncol 2013; 31: 2783-2790.
- Abd El-Rehim DM, Ball G, Pinder SE, Rakha E, Paish C, Robertson JF, Macmillan D, Blamey RW and Ellis IO. High-throughput protein expression analysis using tissue microarray technology of a large well-characterised series identi es biologically distinct classes of breast cancer con rming recent cDNA expression analyses. Int J Cancer 2005; 116: 340-350.
- Sotiriou C and Pusztai L. Gene-expression signatures in breast cancer. N Engl J Med 2009; 360: 790-800.
- Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB, van’t Veer LJ and Perou CM. Concor- dance among gene-expression-based predic- tors for breast cancer. N Engl J Med 2006; 355: 560-569.
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y and Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for se- lection of targeted therapies. J Clin Invest 2011; 121: 2750-2767.
- Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD, Datto MB, Kelley M, Mathey- Prevot B, Potti A and Nevins JR. A pathway-based classi cation of human breast cancer. PNAS 2010; 107: 6994-6999.
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samara- jiwa S, Yuan Y, Graf S, Ha G, Haffari G, Basha- shati A, Russell R, McKinney S, Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Puru- shotham A, Borresen-Dale AL, Brenton JD, Ta- vare S, Caldas C and Aparicio S. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Na- ture 2012; 486: 346-352.
- TCGA. 341 authors. Comprehensive molecular portraits of human breast tumours. The Cancer Genome Atlas Network* Nature 2012;490:61-70
- Dai X, Chen A and Bai Z. Integrative investiga- tion on breast cancer in ER, PR and HER2-derived subgroups using mRNA and miRNA expression pro ling. Sci Rep 2014; 4: 6566.
- Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000; 406(6797):747
- van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415(6871):530
- van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347(25):1999
- Hon JDC, Singh B, Sahin A, Du Gang, Wang J, Wang VY, Deng F, Zhang DY, Monaco, ME, Lee P Breast Cancer Molecular Subtypes: from TNBC to QNBC. Am J Cancer Res 2016;6(9):1864-1872
- William F. Anderson, Philip S. Rosenberg, Aleix Prat, Charles M. Perou, Mark E. Sherman How Many Etiological Subtypes of Breast Cancer: Two, Three, Four, or More? J Natl Cancer Inst. 2014;106(8)
- Spitale A, Mazzola P, Soldini D, Mazzucchelli L and Bordoni A. Breast cancer classification according to immunohistochemical markers:clinicopathologic features and short-term sur- vival analysis in a population-based study from the South of Switzerland. Ann Oncol 2009; 20: 628-635




